Interfacing PyTorch models with collections of AnnData objects via annbatch#
Author: Sergei Rybakov Author: Ilan Gold
This tutorial demonstrates how to set up a annbatch.Loader for an in-memory AnnData object to interface with a PyTorch model and lay out complex covariates.
Note, however, that when training models on many large AnnData objects, loading them all into memory at once is impractical as this notebook does.
annbatch.Loader solves this by allowing users to streams batches formed of contiguous on-memory runs of the training data across multiple AnnData objects without requiring a full in-memory concatenation — compatible with PyTorch, JAX, and other frameworks while being extremely fast.
Preshuffling is a crucial part of making this “contiguous fetching” strategy work - see annbatch.DatasetCollection for more information on performing the on-disk, lazy preshuffle.
To install annbatch for in-memory sparse data (which is most common in single-cell RNA-seq and will be used in this tutorial), use pip install "annbatch[numba]" - note that the numba extra is required for in-memory sparse data.
To use the preload_to_gpu option in annbatch.Loader, install the cupy-cudaxx extra as well where xx is your cuda version.
See the docs for more info on installation.
import torch
import torch.nn as nn
import pyro
import pyro.distributions as dist
import numpy as np
import scanpy as sc
import pandas as pd
from sklearn.preprocessing import OneHotEncoder
from annbatch import Loader
import anndata as ad
import pooch
pyro.clear_param_store()
VAE model definition#
The task is semi-supervised label transfer: pbmc cells have annotated cell types, while covid cells do not.
The model learns a shared latent space across both datasets and uses the labeled pbmc cells to predict cell types for the unlabeled covid cells.
Batch labels (study of origin) are included as covariates to correct for dataset-specific effects.
Create a concatenated AnnData from two AnnData objects#
The data is from this scvi reproducibility notebook.
download("https://scverse-exampledata.s3.eu-west-1.amazonaws.com/anndata/cite_covid_full.h5ad", "covid_cite.h5ad", "cb9745b2d642459926194961f34110a17c16ef6b11777304a3478e12bd682657")
download("https://scverse-exampledata.s3.eu-west-1.amazonaws.com/anndata/pbmc_seurat_v4.h5ad", "pbmc_seurat_v4.h5ad", "c3b0100a6ce27beb64eff53692e09f98da2a58cfdfea08d15ff204f834b41396")
covid = ad.read_h5ad('covid_cite.h5ad')
pbmc = ad.read_h5ad('pbmc_seurat_v4.h5ad')
covid.obs['size_factors'] = np.asarray(covid.X.sum(1)).ravel()
pbmc.obs['size_factors'] = np.asarray(pbmc.X.sum(1)).ravel()
We select highly variable genes using pbmc as the reference, since it has rich cell type annotations and multiple batches to compute variability across.
Concatenating with join='inner' then restricts covid to the same gene set.
sc.pp.highly_variable_genes(
pbmc,
n_top_genes=4000,
flavor="seurat_v3",
batch_key="orig.ident",
subset=True,
)
covid
AnnData object with n_obs × n_vars = 57669 × 33538
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'RNA_snn_res.0.4', 'seurat_clusters', 'set', 'Resp', 'disease', 'subj_code', 'covidpt_orhealth', 'mito', 'ncount', 'nfeat', 'bust_21', 'og_clust', 'severmod_other', 'og_clusts', 'nCount_ADT', 'nFeature_ADT', 'UMAP1', 'UMAP2', 'final_clust', 'final_clust_v2', 'new_pt_id', 'Resp_og', 'final_clust_withnum', 'final_clust_review', 'Age', 'Gender', 'Gender_num', 'size_factors'
obsm: 'pro_exp'
pbmc
AnnData object with n_obs × n_vars = 161764 × 4000
obs: 'nCount_ADT', 'nFeature_ADT', 'nCount_RNA', 'nFeature_RNA', 'orig.ident', 'lane', 'donor', 'time', 'celltype.l1', 'celltype.l2', 'celltype.l3', 'Phase', 'nCount_SCT', 'nFeature_SCT', 'X_index', 'size_factors'
var: 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'hvg'
obsm: 'protein_counts'
dataset = ad.concat(
{'covid': covid, 'pbmc':pbmc},
join='inner',
label='dataset',
)
dataset
AnnData object with n_obs × n_vars = 219433 × 4000
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_ADT', 'nFeature_ADT', 'size_factors', 'dataset'
Setup important covariates in the concatenated object as well as one-hot encoders.
dataset.obs['orig.ident'] = dataset.obs['orig.ident'].astype(str)
dataset.obs.loc[covid.obs_names, 'orig.ident'] = covid.obs['set']
dataset.obs['orig.ident'] = dataset.obs['orig.ident'].astype("category")
dataset.obs['size_factors'] = dataset.X.sum(1)
labels = 'celltype.l1'
dataset.obs[labels] = pd.concat([pd.Series(index=covid.obs_names, data=["not-annotated"] * covid.shape[0]), pbmc.obs["celltype.l1"].astype("str")]).astype("category")
dataset.obs[labels]
AAACCCACACCAGCGT-1 not-annotated
AAACCCACATCTCAAG-1 not-annotated
AAACGAAAGACCTGGA-1 not-annotated
AAACGCTCAGTGGGTA-1 not-annotated
AAACGCTGTAGCTTGT-1 not-annotated
...
E2L8_TTTGTTGGTCGTGATT CD8 T
E2L8_TTTGTTGGTGTGCCTG Mono
E2L8_TTTGTTGGTTAGTTCG B
E2L8_TTTGTTGGTTGGCTAT Mono
E2L8_TTTGTTGTCTCATGGA Mono
Name: celltype.l1, Length: 219433, dtype: category
Categories (9, object): ['B', 'CD4 T', 'CD8 T', 'DC', ..., 'NK', 'not-annotated', 'other', 'other T']
use_cuda = torch.cuda.is_available()
use_cuda
True
encoder_study = OneHotEncoder(sparse_output=False, dtype=np.float32)
encoder_study.fit(dataset.obs['orig.ident'].to_numpy()[: , None])
encoder_celltype = OneHotEncoder(sparse_output=False, dtype=np.float32)
encoder_celltype.fit(pbmc.obs[labels].to_numpy()[: , None])
Loader and model initialization#
We initialize a annbatch.Loader from the concatenated dataset.
chunk_size: number of AnnData chunks streamed at a time. For in-memory data you can safely choosechunk_size=1for perfect random sampling; for on-disk data, larger values with pre-shuffled data are important for maximizing both training quality and speed.preload_nchunks: how many chunks to buffer before yielding batches - here it can be the same asbatch_sizebecause we havechunk_size=1.batch_size: number of cells per training batch. Typical values are 256–8192; larger batches train faster but require more GPU memory.preload_to_gpu: reads directly into pinned GPU memory for a speed boost; requires thecupy-cudaxxextra.
loader = Loader(chunk_size=1, preload_nchunks=8192, batch_size=8192, shuffle=True, preload_to_gpu=True).add_adata(dataset)
pyro.clear_param_store()
torch.set_float32_matmul_precision("high")
n_conds = len(dataset.obs["orig.ident"].values.categories)
n_classes = len(dataset.obs[labels].values.categories) - 1
latent_dim = 20
cvae = CVAE(dataset.shape[1], n_conds=n_conds, n_classes=n_classes, hidden_dims=[1280, 256], latent_dim=latent_dim)
if use_cuda:
cvae.cuda()
Train the model#
optimizer = pyro.optim.Adam({"lr": 1e-3})
svi_sup = pyro.infer.SVI(cvae.model, cvae.guide,
optimizer, loss=pyro.infer.TraceMeanField_ELBO())
# very slow for sequential
svi_unsup = pyro.infer.SVI(cvae.model, pyro.infer.config_enumerate(cvae.guide, 'parallel', expand=True),
optimizer, loss=pyro.infer.TraceEnum_ELBO())
def train(svi_sup, svi_unsup, train_loader):
epoch_loss = 0.
for batch in train_loader:
# select indices of labelled cells
sup_idx = (batch["obs"]["dataset"] == "pbmc").to_numpy()
X = batch["X"].to_dense().int()
cols = ['orig.ident', labels, 'size_factors']
obs_dict = batch["obs"][cols].to_dict("series")
obs_dict['size_factors'] = torch.Tensor(obs_dict['size_factors'].to_numpy()).cuda()
obs_dict['orig.ident'] = torch.Tensor(encoder_study.transform(obs_dict['orig.ident'].to_numpy()[: , None])).cuda()
# do supervised step for the labelled cells
sup_data = {
"orig.ident": obs_dict["orig.ident"][sup_idx],
labels: torch.Tensor(
encoder_celltype.transform(
obs_dict[labels].loc[sup_idx].to_numpy()[:, None]
)
).cuda(),
"size_factors": obs_dict["size_factors"][sup_idx],
}
sup_data['supervised'] = True
epoch_loss += svi_sup.step(X[sup_idx], *sup_data.values())
# do unsupervised step for unlabelled cells
unsup_data = {
"orig.ident": obs_dict["orig.ident"][~sup_idx],
labels: torch.empty(((~sup_idx).sum(), n_classes)).cuda(),
"size_factors": obs_dict["size_factors"][~sup_idx],
}
unsup_data['supervised'] = False
epoch_loss += svi_unsup.step(X[~sup_idx], *unsup_data.values())
normalizer_train = train_loader.n_obs
total_epoch_loss_train = epoch_loss / normalizer_train
return total_epoch_loss_train
NUM_EPOCHS = 250
for epoch in range(NUM_EPOCHS):
total_epoch_loss_train = train(svi_sup, svi_unsup, loader)
if epoch % 40 == 0 or epoch == NUM_EPOCHS-1:
print("[epoch %03d] average training loss: %.4f" % (epoch, total_epoch_loss_train))
Training is complete. We now run inference over the full dataset to extract latent representations and transfer cell type labels from pbmc to the unlabeled covid cells.
Check the results#
Get means of latent variables and cell labels predictions for the unlabelled data.
dataset.obsm['X_cvae'] = np.empty((dataset.shape[0], latent_dim), dtype='float32')
dataset.obs['cell_type_pred'] = pd.Series(dtype=pbmc.obs[labels].dtype)
loader = Loader(chunk_size=512, preload_nchunks=8192, batch_size=8192, shuffle=False, preload_to_gpu=True, return_index=True).add_adata(dataset)
for batch in loader:
latents = cvae.encoder(batch["X"].to_dense(), torch.Tensor(encoder_study.transform(batch["obs"]['orig.ident'].to_numpy()[: , None])).cuda())[:, :latent_dim].detach()
predict = cvae.classifier(latents).detach()
one_hot = torch.zeros(predict.shape[0], predict.shape[1], device=predict.device)
one_hot = one_hot.scatter_(1, predict.argmax(dim=-1, keepdim=True) , 1.).cpu().numpy()
dataset.obsm['X_cvae'][batch["index"]] = latents.cpu().numpy()
dataset.obs.iloc[batch["index"], 8] = np.ravel(encoder_celltype.inverse_transform(one_hot))
dataset.obs['cell_type_joint'] = dataset.obs['cell_type_pred']
dataset.obs.loc[pbmc.obs_names, 'cell_type_joint']=pbmc.obs[labels]
sc.pp.neighbors(dataset, use_rep='X_cvae')
sc.tl.umap(dataset)
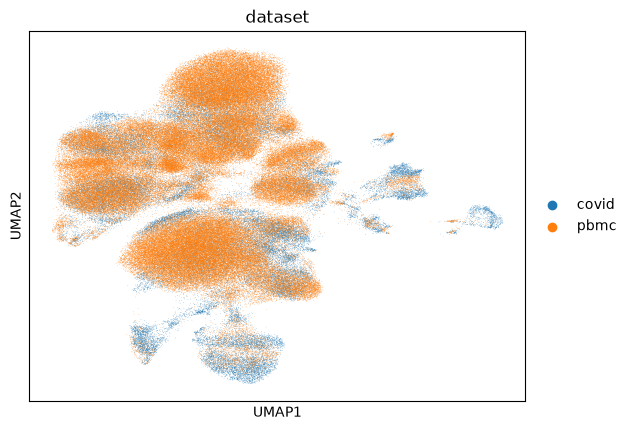
sc.pl.umap(dataset, color=['dataset'])
accuracy = (dataset.obs['cell_type_pred'].loc[pbmc.obs_names]==pbmc.obs[labels]).sum().item()/pbmc.n_obs
accuracy
0.9397764644791178
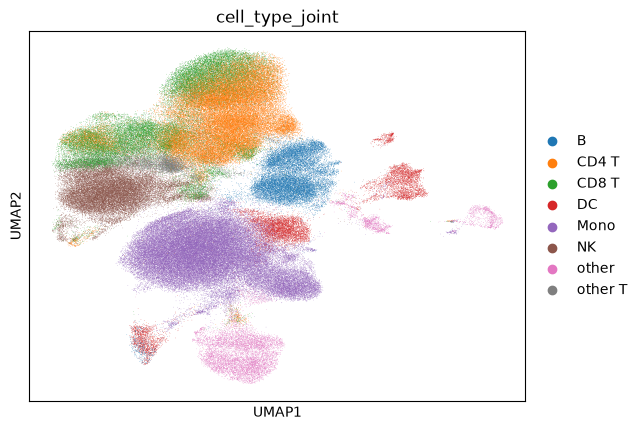
The model achieves ~94% accuracy on pbmc cell types, confirming that the shared latent space captures biologically meaningful structure. Below we visualize the joint UMAP colored by predicted cell type, which also covers the unlabeled covid cells.
sc.pl.umap(dataset, color='cell_type_joint')